Gene therapy for genetic diseases using CRISPR is aimed not only at reducing symptoms, but at changing the molecular cause of the disease. The name CRISPR is also pronounced as ...

15Jul

AMT-130 gene therapy for Huntington’s disease has become one of the most discussed topics in neurology, biotechnology, and clinical research. It is not a drug for temporary symptom control but an experimental technology aimed at addressing the root cause of the disease – the pathological activity of the HTT gene.
Huntington’s disease is a hereditary neurodegenerative disorder characterized by progressive deterioration in movement, cognition, behavior, speech, working capacity, and self-care. The cause is linked to an expansion of CAG repeats in the HTT gene, leading to accumulation of the abnormal huntingtin protein in the brain.
Key search queries expressing the audience’s interest focus on diagnosis, prognosis, and new treatment methods. Therefore, AMT-130 is often searched as “new treatment for Huntington’s disease,” “Huntington’s gene therapy,” “uniQure AMT-130,” “AMT-130 results,” “HTT treatment,” “drug for Huntington’s disease,” and “clinical trials for Huntington’s disease.”
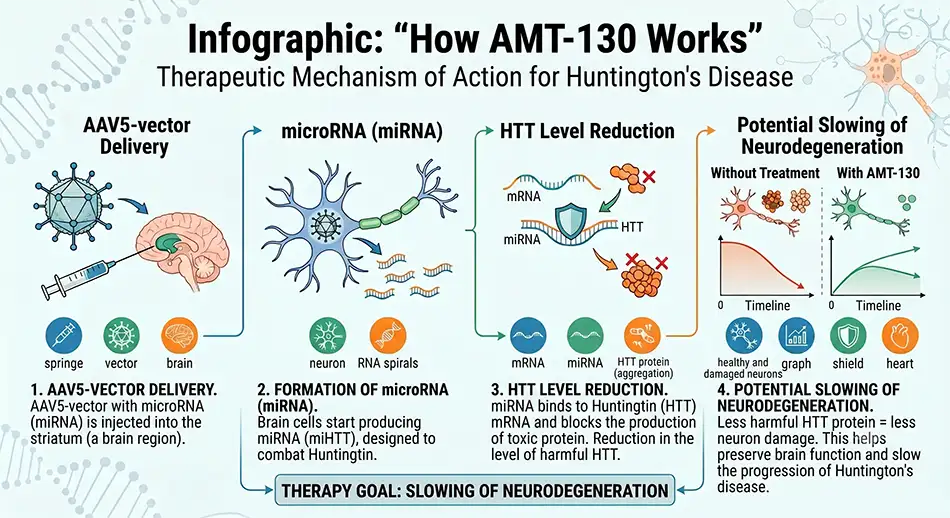
AMT-130 is developed by uniQure as a one-time therapy delivered directly to brain regions most affected by Huntington’s disease. At the core of the technology is an AAV5 viral vector and a microRNA designed to reduce the production of the toxic huntingtin protein.
The mechanism of action can be explained by the sequence of events following therapy administration:
This approach distinguishes AMT-130 from symptomatic medications used to treat chorea, depression, anxiety, sleep disturbances, or behavioral changes. The focus is not on individual symptoms but on the disease-causing mechanism.
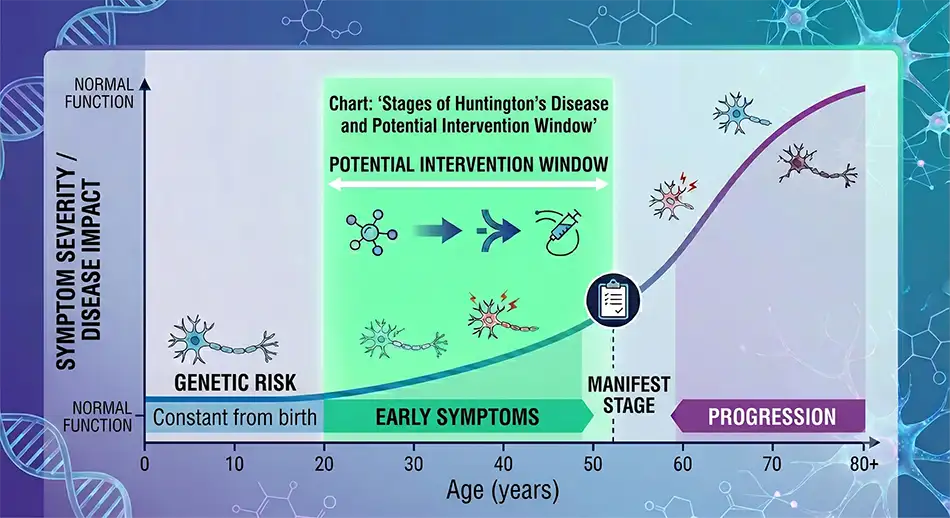
Huntington’s disease is inherited in an autosomal dominant manner. If one parent carries the pathogenic gene variant, the child has a 50% risk of inheriting it. In adult patients, the disease usually manifests during working age, while juvenile forms tend to have a more aggressive course.
For patients and families, the most distressing aspects are not just movement symptoms. The disease gradually affects personality, memory, planning, emotional control, and social independence.
Commonly described symptoms of Huntington’s disease include:
The combination of motor, cognitive, and psychiatric symptoms makes the search for therapies that slow progression critically important. AMT-130 attracts attention because it targets an early stage of the pathological process.
In 2025, uniQure reported 36-month results from the Phase I/II AMT-130 trial. High-dose data became a breakthrough for patients, clinicians, and the biotech market.
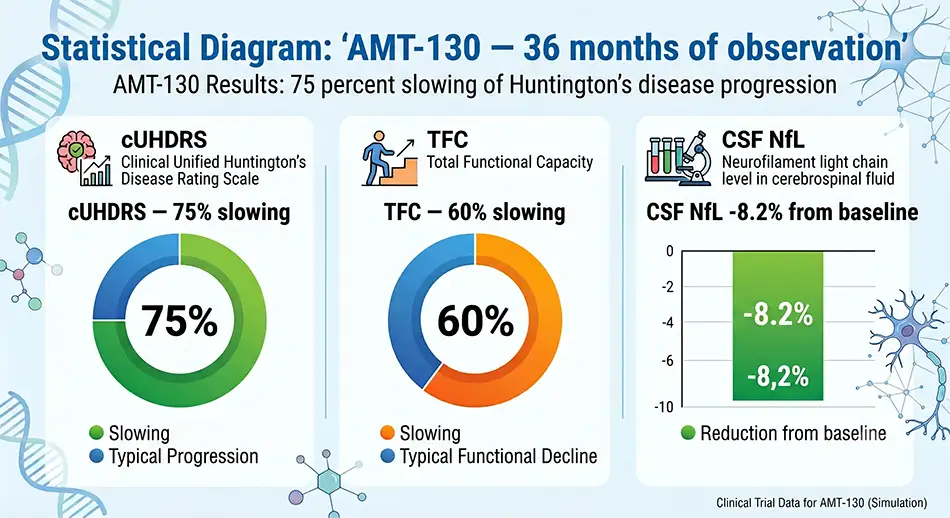
Reported outcomes in the high-dose AMT-130 group included:
These numbers do not indicate a cure for Huntington’s disease. Rather, they show slower functional loss in the treated group compared to an external control dataset.
In 2025, AMT-130 was granted Breakthrough Therapy designation by the FDA. Previously, it also received RMAT, Orphan Drug, and Fast Track statuses. These designations do not approve market sales but may expedite regulator interactions if preliminary clinical data show potential benefits.
In March 2026, the regulatory situation became more complex. The FDA did not agree that Phase I/II data compared to external controls were sufficient as the primary proof of efficacy for marketing approval. The agency recommended conducting a prospective, randomized, double-blind trial with sham surgery controls.
This decision changed the tone of news around AMT-130. While headlines in September 2025 focused on “75% slowing of Huntington’s disease,” in 2026, key searches shifted to “AMT-130 FDA,” “AMT-130 approval,” “AMT-130 Phase 3,” “AMT-130 clinical trial update,” and “is AMT-130 approved.”
For brain gene therapy, controlled trials are more complex than for oral drugs. AMT-130 is surgically delivered into the striatum—the caudate nucleus and putamen. Hence, the regulatory standard raises ethical, medical, and practical debates.
Sham surgery control involves mimicking the surgical procedure in the control group to compare the therapy effect with the placebo effect, procedural impact, and natural disease progression. This is particularly sensitive for patients with progressive neurodegenerative conditions.
Main risks and issues of this study design include:
This does not cancel out scientific interest in AMT-130 but shows that the path of gene therapies to patients depends not only on biological logic and initial results but crucially on study design, evidence quality, safety, long-term follow-up, and FDA stance.
AMT-130 is studied in patients with early manifest Huntington’s disease. The early stage is critical since therapy aimed at reducing toxic protein theoretically makes more sense before massive neuron loss.
Patients and families commonly ask:
There are no simple commercial answers yet. AMT-130 remains experimental, not a standard treatment. Access, participation criteria, dosage, risks, neurosurgical procedure, and long-term follow-up are determined by clinical trial protocols and regulatory decisions.
Current Huntington’s disease treatment primarily focuses on symptom management, rehabilitation, psychiatric support, nutrition, speech therapy, physical therapy, and family support. Gene therapy changes the conversation from “how to ease symptoms” to “can we slow the biological process?”
In this context, AMT-130 competes not only with other genetic approaches but also with a broader class of therapies targeting HTT, RNA, protein aggregates, neuroinflammation, and neurodegeneration biomarkers. Patients should understand that strong interim results do not equal automatic approval, and Breakthrough Therapy status does not guarantee market availability.
Key facts about AMT-130 as of 2026:
The story of AMT-130 has been a test for the entire gene therapy field in neurodegenerative diseases. It showed that even very promising clinical signals must withstand rigorous evidence evaluation, especially when delivered into the brain and claiming to alter an incurable disease’s course.






